As a deal maker, where should I go for a deal? Where is my competition?
There is so much written about China, I thought I would try to put it in the context of other countries.
DEMOGRAPHICS: China is Big but low GDP per capita, Japan has the oldest population. Both Japan and China may have reached peak population, while the US has immigration to continue growth. China and Japan have more big cities (making clinical trial recruitment easier).
The Medical Culture varies tremendously.
The US, with the 3rd largest population and private insurance, has the biggest market
But there are even bigger differences in the magnitude of sales of new drugs. In the US, to be in the top 10 in 2031 means double digit billions.
The biggest company R&D budgets per company are in “Global companies”.
The biggest European and Japanese companies have become global companies.
The biggest companies in the US have 45%-70% of their Rx sales in the US.
The biggest companies in Europe have 15%-30% of their Rx sales in Europe.
The biggest companies in Japan have <10% (Takeda) to 39% of their sales in Japan. (Smaller Japanese companies have most of their Rx sales in Japan)
The biggest Chinese companies have 80-95% of their sales in China.
The biggest Korean companies with biologics (Samsung and Celltrion) have 10-20% of their sales in Korea. The other big Korean companies have 70-90% of their Rx sales in Korea.
But China has almost as many drugs in Phase 1 thru 3 as the US, in almost as many companies as in the US.
There are more companies getting series A in the US and in China but the dollar amount is smaller in China.
The US leads in IPOs
But the Hong Kong Hang Seng Biotech Index was up 87% year to date (while the US XBI was down 6%).
Europe is active in company acquisitions, but Asia is not.
For companies with headquarters in the US, Europe, Japan and Korea: most partnering deals are early and with more in-licensing than out-licensing.
China does more out-licensing than in-licensing.
So as a deal-maker, what do I think this data suggests?
1) You need to capture value from the US, the biggest market and home of blockbusters.
2) US companies do the most in-licensing. US and Europe do the most M&A.
3) The most deals in 2024 and 1st Half of 2025 are still done at discovery and preclinical.
4) Japanese companies are increasingly global companies and do more in-licensing than out-licensing.
5) China is a source of drugs to bring in, with many drugs in the pipeline and new series A companies needing partners to maximize their value. China does more out-licensing than in-licensing. Presumably, the huge China vs China competition is pushing Chinese companies to innovate more to compete and to do deals. And more exits (IPOs and M&A) encourages more VC funding of innovation.
6) But the low cost and the high populations cities (for fast recruitment) means China should be considered for collaborations for your drug development. (Just remember you need 20% of patients in the US for FDA approval).
7) Korea is a high-income market but small. In-licensing deals are often early or at market stage.
As the saying goes, “What’s in a name? That which we call a Series A by any other name would smell as sweet.” Er… something like that, right? Hmmm, maybe it went a little bit differently.
But whatever it be, or not to be😊, the Seed Round is the new Series A. Clearly. I think we’ve all felt it for sometime but the data is in and the good ‘ole Series A just don’t buy what it used to. Nahhh… the Seed round does that, and it may buy more (equity) than it used to as a Series A (more data hunting and crunching required but one gets a sense that the venture capitalists are, well, capitalizing).
Labiotech does a really nice job collecting and summarizing a variety of topics related to financings and dealmaking in the biotech sector and the 2024 breakdown of funding offers the following approximations (roughly, with some rounding made by this author):
The internal breakdowns for amounts invested look like this:
Readers of this corner will know that we keep a close eye on the XBI
As usual, the outliers can skew the numbers (more on this in a moment) but the median amounts invested into these rounds puh-rihhhty much drive the nail in the coffin of the old thinking about Series dynamics. This data could be charted in another way in which an inverted bell curve would appear and a GAPING hole between $20M and $50M would stare back at you. Think about that for a moment… if you can’t get to value inflection for ~$15-20M, you better be raising $60-75M and have multiple reasons to do so as a cursory view of the companies listed in the dataset further indicates that the lower outliers (sub-median) on the Series A were generally geared for “finding out” about a single asset in the clinic.
Back to that previously mentioned outlier that can skew the averages… it also happens to bring even more of a spotlight to those famed words from Shakespeare which began this Corner on Market Sentiments. One of the companies in the 2024 data set raised a whopping $100,000,000 … as a Seed Round!! Indeed, a rose by any other name…
Molecular glue degraders (MGDs) are currently having a bit of a moment. In the first half of 2025, the number of papers describing such compounds has doubled.
Molecular glue uptick in the literature —a doubling of output since the start of the year. (Search based on number of papers indexed in PubMed with search term “molecular glue” in title or abstract. As of June 30, a total of 28 preprints describing molecular glue drugs had been published on BioRxiv/MedRxiv.)
2025 has also witnessed a whole raft of MGD startups publish research related to their programs:
Startup (location)
Scientific founders (location)
2025 paper
Ambagon Therapeutics (Eindhoven, The Netherlands)
Michelle Arkin (UCSF, San Francisco, CA), Luc Brunsveld and Christian Ottman (Eindhoven University of Technology)
Rajesh Chopra and Ian Collins (The Institute of Cancer Research and Cancer Research UK); Nicolas Thomä (Friedrich Miescher Institute, Basel, Switzerland)
Héctor G. Palmer, Esther Riambau, Isabel Puig, Josep Tabernero, Xavier Barril, and Carles Galdeano (Vall d’Hebron Institute of Oncology, University of Barcelona and ICREA)
On the commercial front, the march of startups receiving funding shows no sign of slowing down, with Trimtech Therapeutics and Booster Therapeutics raising substantive rounds. The first few months of the year have also seen the continuation of last year’s pharma MGD scramble to license programs from Triana Biomedicines and Neomorph, with deals based around molecular glues from Abbvie and Merck targeting Neomorph and Springworks, respectively.
Unlike their more recent cousins, the PROTACs (proteolysis targeting chimeras), MGDs have a long history. The archetypal MGD, thalidomide, was discovered back in the 1950s. From the late 1990s, a new generation of immunomodulatory imide drug (IMiD) derivatives of thalidomide were synthesized, culminating with the approvals of lenalidomide and pomalidomide for myeloma (which formed the basis for the Celgene (now BMS) franchise).
Unlike PROTACs, which use two ligands with a linker and tend to be rather unwieldy, MGDs are small, single compounds that induce conformational changes in E3 ubiquitin ligases and target proteins, reshaping both to enable binding. The vast majority of MGDs bind Cereblon (CRBN), leading to ubiquitination of the protein of interest and degradation in the 26S proteasome, although work is progressing to broaden MGD action to some of the other 600 or so E3 ubiquitin ligases (e.g., DCAF11,15 or 16, DDB1, SIAH, KEAP1, VHL, β-TrCP, Nedd1 and, just last week, TRIM21).
A key challenge in finding new MGDs has been a lack of understanding of the structural rules whereby MGDs turn their target proteins into CRBN ‘neosubstrates’, which has meant MGD ‘hit-finding’ is much more challenging, with fewer degrees of freedom than PROTACs.
What drug hunters have established is that many protein targets of glues contain a β-hairpin structural motif known as the ‘G-loop’. When a MGD brings a target together with CRBN, one end of the MGD interacts with a binding pocket in the C-terminal domain of CRBN, while the other end protrudes from the pocket and interacts with the G-loop (part of the so-called ‘degron’) in the neosubstrate. But how many proteins possess the β-hairpin G-loop or whether the loop is strictly necessary for MGD action have remained open questions. A recent study by Monte Rosa Therapeutics’ scientists starts to tackle these issues, disclosing a large cadre of potential new substrates for CRBN, some of which depart from the canonical β-hairpin G-loop, radically expanding MGD target space.
To map the full range of proteins potentially recruitable by CRBN through MGDs, the team led by John Castle and Sharon Townson developed computational algorithms to search for β-hairpin G-loop motifs in protein structures from two databases: Protein Data Bank and AlphaFold2. This approach resulted in 1424 candidate proteins, some of which were experimentally validated in MGD assays. The list included previously known neosubstrates, but also new proteins such as NEK7—a protein of interest as an autoimmunity target.
The researchers then wondered if the full β-hairpin structure of the G-loop is required for CRBN recognition and rescreened the structure databases looking for a minimal, structurally defined helical G-loop motif. This resulted in the identification of 184 additional potential neosubstrates, including mTOR, a well-established therapeutic target for drugs like rapamycin and sirolimus. Crystallographic data showed that the binding of this helical G-loop to CRBN is similar to that of the canonical β-hairpin G-loops.
As these protein–protein interactions have been well characterized, the team then tried to identify an even wider set of potential neosubstrates, looking now for proteins with sequences that might result in surfaces with electrostatic properties similar to known CRBN interactors, independently of secondary structure and the existence of G-loops. Using surface-matching algorithms, they identified and validated VAV1 (another autoimmune disease target) as a CRBN neosubstrate, providing compelling evidence that G-loops are not strictly necessary for the action of MGDs.
These findings show that CRBN recruitment through MGDs can be driven by a broader set of structural features than previously thought. The identification of a large number of neosubstrates potentially opens up a whole new set of previously ‘undruggable’ targets to MGDs (>1,600 proteins from many target classes, according to the Monte Rosa team).
An expanding protein target universe for MGD drugs. Source: Science
The big questions, though, are still ahead. How will drug developers mitigate the risks of ‘off-tissue’ toxicity as this swathe of novel MGD compounds and new targets make their way into the clinic?One answer to the toxicity concern is molecular glue antibody conjugates (MACs), which can better localize glues to the tissue of interest. But that’s a subject for a whole other future Haystack Chat!
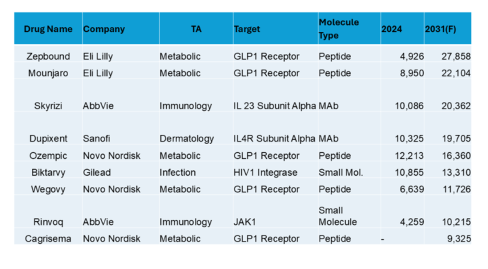
What do analysts think will be the top 10 drugs in the year 2031 (as searched in GlobalData)?
Top 10 in the US in 2031 (USD Millions)
In the US analyst forecasts for 2031, obesity dominates (with immunology, derm and infection for other TAs). Along with the obesity peptides are 2 small molecules and 2 MAbs. The sales are in the tens of billions.
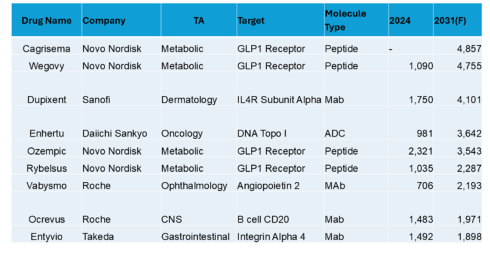
Top 10 in Europe (USD Millions)
In Europe, along with the obesity drugs and dupixent in derm, we see the oncology ADC Enhertu, the ang2 ophthalmology drug Vabysmo, a CNS CD20, and a GI integrin in the top 10. Sales are in the single digit billions.
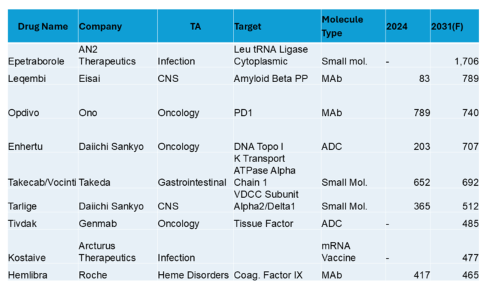
Top 10 in Japan (USD Millions)
In Japan, obesity is not visible in the top 10. An anti-infective tops the list, followed by CNS, oncology, GI and including heme disorders. There are companies not in the top 20 for global sales. Most of the top 10 have sales below $1B.
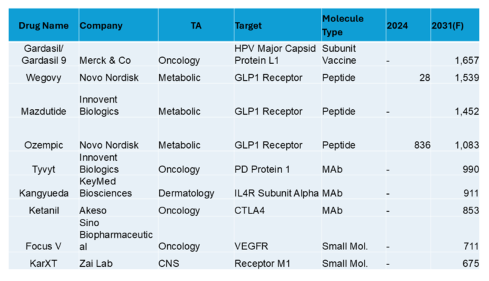
Top 10 in China (USD Millions)
For China, obesity is back in the top 10, but Gardasil, an oncology HPV vaccine tops the list. Local company “fast followers” are apparent and most of the top forecasted drugs are not yet launched (presumably a reflection of the rapidly evolving pharmaceutical environment). To get into the top 10, sales are above $500M.
Conclusion: The marketplace for drugs shows considerable variation in different regions around the world.
Just over a week ago, AbbVie paid $2.1 billion for Capstan Therapeutics’ in vivo anti-CD19 chimeric antigen receptor (CAR)-T cell therapy (CPTX2309) for B cell-mediated autoimmune disorders, which is currently in phase 1 testing. In the past few days, EsoBiotec (acquired by AstraZeneca earlier in the year) also published its first clinical data on a lentiviral-delivered anti-B-cell maturation antigen (BCMA) CAR-T approach (ESO-T01) for multiple myeloma, detailing responses in four patients, two of whom showed complete remission. With a host of other companies working on in vivo delivery into endogenous T cells—including Interius BioTherapeutics, Umoja Biopharma, and Orna Therapeutics, the field of in vivo delivered CAR-T cells appears poised at a tipping point.
Ex vivo generation of CAR-T cells (left) is a complex and lengthy procedure, entailing isolation of T cells from patient blood (1), followed by activation, transduction, and ex vivo expansion for several weeks. After undergoing conditioning treatment (2), patients are infused with a bolus of expanded CAR-T cells (3). In the in vivo approach (right), delivery vector (targeted LNPs or lentiviruses, depicted as red dots) are infused directly into the patient, where they encounter T cells and selectively deliver genetic material encoding the CAR (red). Source: Molecular Therapy.
Since transforming the face of cancer treatment in 2017, autologous CAR-T cell therapy has been dogged by logistical issues that have limited commercial rollout and increased costs—the need for leukapheresis, laborious cell harvesting, heterogeneous cell expansion, lengthy turnaround times, and inconsistency of batches—with access limited to just a few clinical centers. Extensive waiting lists can mean many patients die before even being treated, which has driven the search for ex vivo approaches that shorten manufacturing times using fully closed systems and/or miniaturization. Given these challenges, delivery of a CAR-encoding mRNA to a T cell in vivo could be a game-changing technology: No need for viral vectors; no leukapheresis/chemo; no ex vivo manipulation, no requirement for multiple patient hospital visits; no convoluted training of personnel; and no risk of second primary T-cell cancers due to insertional mutagenesis. This last issue has loomed over the field, with all CAR-T therapies carrying black box warnings, although at the end of June the FDA removed all requirements for Risk Evaluation and Mitigation Strategies (REMS).
Writing in Science, the founding team of Capstan Therapeutics, headed by Carl June and Bruce Levine at the University of Pennsylvania and Haig Aghajanian of Capstan, report proof of concept data that functional CAR T cells with antitumor activity can be produced in animal models without any ex vivo manipulation. A key breakthrough in their effort was the development of lipid nanoparticles (LNPs) specifically designed to target T cells and to overcome the propensity of LNPs to accumulate in the liver. To avoid this problem, the authors screened a set of ionizable lipids to identify L829, a lipid that incorporates a tertiary amine headgroup that reduces non-specific interactions with the hepatic system due to its pH-dependent protonation and neutral charge. Ester cleavage sites in the lipid also promote rapid breakdown in, and clearance from, hepatocytes. A final step was to decorate L829 LNPs with a mAb targeting CD5, a T-cell specific marker. The resulting LNP showed limited liver uptake in rodents and non-human primates compared with control LNPs.
To test the potential of L829-containing LNPs to generate functional CAR-T cells, the team engineered them to incorporate 1) mRNA encoding a CAR that binds CD19 on B cells and 2) an antibody targeting CD8+ T cells. These CD8-L829-CD19 targeted (t)LNPs successfully delivered the mRNA in vitro to CD8+ T cells from healthy subjects and from people with B cell-mediated autoimmune diseases. In vivo, these CAR T cells had anti-tumor activity in a humanized mouse model of B cell acute lymphoblastic leukemia.
The Capstan approach to CAR-T cell therapy: An IV bag, a targeted LNP, and an mRNA encoding the CAR of interest. Source: Science.
In cynomolgus monkeys that received repeated doses of CD8-L829 tLNPs containing anti-CD20 CAR mRNA (instead of anti-CD19, which is not cross-reactive between human and monkey), sustained B-cell depletion was observed that lasted for one month. Importantly, reconstituted B cells were predominantly naïve, implying an immune reset — a key therapeutic goal in autoimmunity.
The Capstan in vivo mRNA-encoded CAR T platform eliminates the need for ex vivo manipulation and lymphodepleting conditioning. It avoids the risks often associated with the use of viral vectors that integrate into the genome. It also is transient, allowing dosages to be optimized and quickly stopped if patients suffer adverse events associated with neurotoxicity or cytokine-release syndrome. It will be interesting to see whether the approach is scalable and whether it can open up conditions where long-term CAR-T cell persistence might not be necessary, such as autoimmune disease.
Going forward, an important question will be to determine the potential immunogenicity of the tLNP formulation (especially as the mRNA treatment may be given multiple times), and whether tLNPs cause elevations of human liver enzymes like alanine transaminase or aspartate aminotransferase. Liver toxicity of a novel liposome formulation already caused a clinical hold for Verve Therapeutics’ base editing therapy last year. Future work will also need to define optimal dosing, durability, and long-term safety of this approach. But the work of June, Aghajanian and their colleagues is a compelling advance promising a new era of widely available adoptive T-cell therapies for B-cell driven hematological cancers and autoimmune conditions. A single dose of any of the seven currently approved commercial ex vivo CAR-T therapies costs ~$500,000. A vial of an in vivo treatment is likely to cost an order of magnitude less.
Acceleration of laboratory-based technical and computational cross-fertilization, and ethical and cost pressures on regulatory bodies and therapeutic innovators is driving advancements in preclinical human-based technologies.
Organ (Lab)-on-chip (OoC/LoC)is one of the most striking examples of new translational research technology expansion with ~35% CAGR expected over the next decade (below).
Collaborations between academia and CRO’s are driving improvements in organoid technology for the field of oncology broadly and are expected to improve OoC adoption. Academic innovation using commercial OoC technology is also advancing applications in specific indications in oncology. CRO’s continue to build off established uses in ADME and toxicology to explore R&D applications in oncology space and have even combined organ systems to support elaboration of multiple drug parameters in a single assay.
DEALS
The Tara Biosystems – Valo Health deal is a nice example of how an organ-on-a-chip technology approach has driven collaborations, acquisitions and deals:
Tara Biosystems and GSK collaborate on CV drug profiling (2019)
Valo Health acquires Tara Biosystems for CV OoC platform (2022, ~$75M upfront)
Valo and Novo Nordisk sign CV drug discovery deal (2023, $60M upfront, $2.7B total)
Emulate, TissUse and Mimetas have also been backed by strong big pharma collaborations (AstraZeneca, Bayer, Roche) and funding rounds.
Drug development efforts targeting the constitutive 26S proteosome have led to the development of several important multiple myeloma (MM) and mantle cell lymphoma treatments, including the first landmark FDA approval of Millennium Pharmaceuticals’ (now Takeda) dipeptide boric acid Velcade (bortezomib) in 2003 and second-generation molecules, such as Amgen/Ono Pharmaceutical’s irreversible inhibitor Kyprolis (carfilzomib) and Takeda’s orally available inhibitor Ninlaro (ixazomib). Second-generation versions of these ‘pan-proteosome’ drugs have longer duration of effect, reduced peripheral neuropathy and increased safety in renally impaired patients, but may cause gastrointestinal and cardiac toxicity. This toxicological profile has shifted attention to developing inhibitors selective for an alternative form of the core 20S proteosome—the immunoproteasome, which processes peptides for presentation to CD8+ T cells in the MHC-I complex and is constitutively expressed only in hematopoietic cells, induced in immune cells stimulated in the presence of IFN-γ, and upregulated in certain cancers like MM.
The β1 subunit (particle components beta subunit 6; PSMB6), β2 subunit (PSMB7), and β5 subunit (PSMB5) found in the constitutive 26S proteasome (left) are replaced in the immunoproteasome (right) by the β1i subunit (low molecular mass polypeptide 2 (LMP2)/PSMB9), β2i subunit (multicatalytic endopeptidase complex-like 1 (MECL-1)/PSMB10), and β5i subunit (LMP7/ PSMB8), respectively. Existing inhibitors and their sites of action are indicated. Adapted from https://bit.ly/4kgmQj9
Currently, Kezar Life Sciences’ is furthest along in development; in April, it completed a phase 2a trial in autoimmune hepatitis of zetomipzomib (KZ-616), a small-molecule that inhibits both the immunoproteasome core particle component beta subunit 8 (PSMB8; LMP7/β5i) and PSMB9 (LMP2/β1i). Merck kGaA (Darmstadt, Germany) is also pushing forward with a phase 1 clinical program of M3258, a small-molecule inhibitor specific for PSMB8 and intended for use in MM (Principia Biopharma’s selective PSMB8 inhibitor was swallowed up by Sanofi in 2020 when the pharma acquired the San Francisco-based biotech’s Bruton’s tyrosine kinase inhibitor program). Elsewhere, Leiden University startup iProtics recently received a €200K grant from the Dutch Biotech Booster to develop selective immunoproteosome inhibitors, while Auburn University spinout Inhiprot (West Lebanon, NH) received SBIR funding to develop a dual PSMB6/PSMB9 inhibitor for MM. Now, a new study reveals immunoproteosome targeting may also have benefits in neuroinflammatory diseases like multiple sclerosis.
The work, published in Cell and led by Catherine Meyer-Schwesinger and Manuel Friese, from University Medical Center Hamburg-Eppendorf, identifies a neuron-intrinsic mechanism of neurodegeneration in multiple sclerosis (MS) driven by the immunoproteasome.
Under healthy conditions, neurons utilize the constitutive proteasome subunit PSMB5 to regulate proteostasis and degrade 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), a potent stimulator of glycolysis. This degradation is key because neurons rely more on the pentose phosphate pathway than on glycolysis to produce antioxidants like NADPH and glutathione for protection against oxidative stress.
However, Meyer-Schwesinger, Friese and their colleagues show that, during neuroinflammation, chronic exposure to interferon-γ leads to the induction of the immunoproteasome in neurons, triggering the replacement of constitutive proteosome PSMB5 (β5c) with PSMB8 (β5i). This subunit swap in neurons reduces proteasomal activity, resulting in accumulation of PFKFB3, which in turn enhances glycolysis, diminishes the activity of the pentose phosphate pathway, and impairs redox homeostasis — conditions that sensitize neurons to oxidative injury and ferroptosis.
Interferon-induced immunoproteasome markedly decreases proteasomal activity in neurons, leading to a switch in neuronal metabolism from oxidative phosphorylation (left) to glycolysis accompanied by oxidative injury and ferroptosis (right). Source: Cell
The team showed that this mechanism was operational in both experimental autoimmune encephalomyelitis (EAE; a mouse model of MS) and brain tissue from MS patients. Moreover, neuron-specific knock-out of Psmb8 or pharmacological inhibition using the small-molecule PSMB8 inhibitor ONX-0914 (originally developed at Onyx Pharmaceuticals/Proteolix) protected neurons in vivo from inflammation-induced damage. Similarly, blocking PFKFB3 with the small-molecule inhibitor PFK-158 or through conditional knockout in neurons reduced disease severity in EAE, prevented neuronal and synaptic loss, and reduced markers of oxidative stress and lipid peroxidation.
It is important to highlight that, unlike cancer or immune cells, neurons do not upregulate PSMB8 in response to a series of MS-related cytokines. So, the neuron-specific effect reported in this study might only become active upon chronic neuroinflammation (i.e. chronic exposure to interferon-γ). Understanding this mechanism might reveal new targets related to the immunoproteosome in the treatment of MS.
This brings us to challenges for translational efforts seeking to develop immunoproteosome inhibitors against MS. Several important neuronal processes, such as synaptic transmission and calcium signaling, are tightly linked to proteasome function; thus, pan-proteosome inhibitors like Velcade could be detrimental to the CNS. The saving grace of approved proteosome inhibitors is that current chemotypes (boronate-based peptides or epoxyketone-based binders) do not cross the blood brain barrier, at least in healthy individuals. Thus, any MS program might need to use intrathecal injection for compounds derived from existing chemical series or engage a medicinal-chemistry effort to design molecules that can breach the BBB and retain potency.
The gambit for immunoproteosome-selective drugs is that they avoid inhibiting constitutive 26S proteosome activity in most tissues (and non-inflammed CNS), which is what makes Velcade and its derivatives so difficult for patients to tolerate; an immunoproteosome inhibitor should therefore have a more favorable safety profile. But so far, immunoproteosome-targeting drugs have had their own share of toxicity problems in the clinic.
Last October, Kezar abandoned its program for zetomipzomib in lupus nephritis after the FDA placed a clinical hold on the trial after 4 patient deaths. The agency placed a second partial hold on the company’s autoimmune hepatitis trial in 24 patients last November due to concerns about steroid control and injection site reactions in 4 patients who were waiting to roll over into the open-label extension arm. Concerns about compromised immune surveillance of acute or latent viral infections due to hobbled antigen processing and presentation would also need to be explored.
In sum, the new work provides strong evidence that the immunoproteosome plays a key role not only in inflammation or infiltration of immune cells, but also in a metabolic switch in neurons which is a key driver of vulnerability in MS. It will be interesting to see whether either targeting immunoproteosome component PSMB8 or taking a completely different tack, blocking PFKFB3, will prove more practical as a neuroprotective strategy in MS.
The firm is focused on therapeutics companies and does not invest in medical devices, diagnostics, or digital health. The firm is open to considering assets of very early stages, even those as early as lead optimization phase. The firm considers various modalities, including antibodies, small molecules, and cell therapy. Currently, the firm is not interested in gene therapy. Indication-wise, the firm is most interested in oncology and autoimmune diseases but has recently looked at fibrotic diseases and certain rare diseases as well.
The firm is opportunistic across all subsectors of healthcare. Within MedTech, the firm is most interested in medical devices, artificial intelligence, robotics, and mobile health. The firm is seeking post-prototype innovations that are FDA cleared or are close to receiving clearance. Within therapeutics, the firm is interested in therapeutics for large disease markets such as oncology, neurology, and metabolic diseases. The firm is open to all modalities with a special interest in immunotherapy and cell therapy.
A strategic investment firm of a large global pharmaceutical makes investments ranging from $5 million to $30 million, acting either as a sole investor or within a syndicate. The firm is open to considering therapeutic opportunities globally, but only if the company is pursuing a market opportunity in the USA and is in dialogue with the US FDA.
The firm is currently looking for new investment opportunities in enterprise software, medical devices, and the healthcare IT space. The firm will invest in 510k devices and healthcare IT companies, and it is very opportunistic in terms of indications. In the past, the firm was active in medical device companies developing dental devices, endovascular innovation devices, and women’s health devices.
A venture capital firm founded in 2005 has multiple offices throughout Asia, New York, and San Diego. The firm has closed its fifth fund in 2017 and is currently raising a sixth fund, which the firm is targeting to be the largest fund to date. The firm continues to actively seek investment opportunities across a […]